
PMDA(医薬品医療機器総合機構 )とは
医療機器業界に参入して、
製造販売業を目指すならPMDAを知らないで済ませる訳にはいきません。
PMDAは「Pharmaceuticals and Medical Devices Agency」の略称です。
「製薬と医療機器の外局」と訳して良いと思います。
PMDAは、平成16年4月1日に設立しました。
法人としては、中期管理目標法人と言う聞いたことがない、
法人になっています。
「中期管理目標法人」とは、独立行政法人の三つの類型の一つです。
公共上の事務等のうち、その特性に照らし、
一定の自主性および自律性を発揮しつつ当該業務を遂行します。
また、その名の通り中期的(3~5年)な視点にたって執行することが求められます。
そのため、国民の需要に的確に対応し、
多様で良質なサービスの提供を通じた公共の利益の増進を推進することを目的とする法人です。
中期目標管理法人の役員および職員の身分は非公務員です。
2017年(平成29)4月1日時点で53法人です。
例えば、
- 国民生活センター
- 郵便貯金・簡易生命保険管理機構
- 国際協力機構(JICA(ジャイカ))
- 国立美術館
- 高齢・障害・求職者雇用支援機構
- 日本貿易振興機構(JETRO(ジェトロ))
- 都市再生機構、環境
再生保全機構などが中期目標管理法人として設立されています。
では、その中の一つ、PMDAについてご説明します。
PMDAとは
以下は、PMDAのサイトにある説明です。太字強調は私によるものです。
独立行政法人医薬品医療機器総合機構(PMDA;Pharmaceuticals and Medical Devices Agency)は、
平成13年に閣議決定された特殊法人等整理合理化計画を受けて、
国立医薬品食品衛生研究所医薬品医療機器審査センター、
医薬品副作用被害救済・研究振興調査機構及び財団法人医療機器センターの一部の業務を統合し、
独立行政法人医薬品医療機器総合機構法に基づいて平成16年4月1日に設立され、
業務を開始しました。PMDAは、医薬品の副作用や生物由来製品を介した感染等による健康被害に対して、
迅速な救済を図り(健康被害救済)、
医薬品や医療機器などの品質、有効性および安全性について、
治験前から承認までを一貫した体制で指導・審査し(承認審査)、
市販後における安全性に関する情報の収集、分析、提供を行う(安全対策)ことを通じて、
国民保健の向上に貢献することを目的としています。
法律の条文のように読み難い文章ですが、
要するに閣議決定を受けて、
3つの組織を統合してできた、
国民の健康と保険のための組織です。
注意しなければならないことは、
医薬品や医療機器を発達させ、
新しい製品を積極的に作って行くための組織ではないと言うことです。
目的はあくまでも、
国民を健康被害から守ることです。
ここを勘違いすると、
医療機器を開発する上で苦労することになります。
極論すれば、
PMDAは、新しい医療機器等求めていません。
これまでに説明した、医療機器における日本の貿易赤字を減らしたいとも思っていません。
これに熱心なのは、経済産業省のプロジェクトです。
PMDAは、規制する側です。
ですから、新規医療機器を企画開発しようと思って、
PMDAに行くと時々憤慨して帰ってくることになります。

少なくとも、
そのようなことに対して、
PMDAは協力的ではありません。
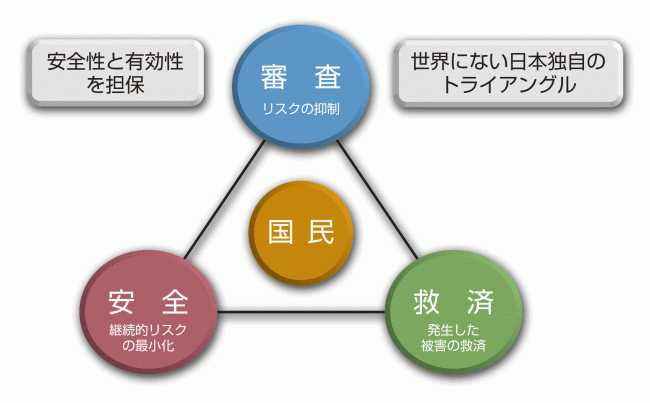
PMDAの主な業務
では、PMDAの主な業務を説明しましょう。
相談業務
PMDAでは、医薬品、医療機器、再生医療等製品の治験や申請資料に対し、
指導・助言を行っています。
これはその通りですが、
親切な対応ではありません。
あまり期待しないことです。
この他、医薬部外品などの簡易相談についても、
申込者に対して指導・助言を行っています。
また、平成23年7月からは、日本発の革新的医薬品、医療機器、再生医療等製品の創出に向けて、
有望なシーズを持つ大学・医療機関、ベンチャー企業を主な対象とした薬事戦略相談を実施してきました。
治験関連業務
治験の実施にあたって、
治験依頼者(製造販売業者等)及び自ら治験を実施する者(医師、歯科医師等)は、
治験計画の届出及び治験中の副作用、不具合等の報告が義務づけられています。
PMDAでは、治験計画届及び治験中の副作用、
不具合等報告の受付等を行っています。
また、治験審査委員会(IRB)についても、
各医療機関からPMDAに登録された情報を公表しています。
ただし、これもどこへ行けば、
治験に対応してくれるかなど、
情報の提供を求めててはいけません。
それは、あくまで自分で行うのです。
PMDAは、それをチェックするだけです。
承認審査業務
承認業務の中には大きく分けて3つの業務があります。
「医薬品等承認審査業務」「医療機器承認審査業務」「再生医療等製品承認審査業務」
この3つの業務です。
順番に見ましょう。
医薬品等承認審査業務
医薬品の承認審査では、薬学、医学、獣医学、理学、生物統計学などの専門知識を有する審査員が、
「品質」「薬理」「薬物動態」「毒性」「臨床」「生物統計」を担当し、
審査チームを形成して審査を行います。
また、審査の過程では、外部専門家との意見交換(専門協議)を行い、
より専門性の高い見地から審査することを目指しています。
また、より優れた医薬品をより早く医療現場に提供するため、
審査期間の目標を設定し、業務の迅速化に取り組んでいます。
さらに、日本、アメリカ、ヨーロッパにおける新医薬品の承認審査資料関連規制の整合化を図ることにより、
データの国際的な相互受け入れを実現することを目的とした医薬品規制調和国際会議(ICH)に参加するとともに、
同会議で合意された内容を積極的に承認審査に取り入れています。
難しいことを言っていますが、
要するに、外国で安全が確認され、
承認された医薬品は比較的早く、
日本でも承認されます。

医薬品等の承認審査では、「新医薬品」のほかに、
「後発医療用医薬品(すでに承認されている医薬品と同一性が認められる医薬品)」、
薬局・薬店で医師の処方せんなしに購入できる「一般用医薬品(OTC)・要指導医薬品」、
その他「医薬部外品」を審査しています。
医療機器承認審査業務
医療機器は、医薬品と同じく、
疾病の診断、治療、予防など、医療に用いる製品という特性があります。
しかし、その他、医療機器独特の問題として、メスやピンセットから、MRI、ペースメーカーまで、
製品ごとに基になる技術・素材が異なり、
使用形態、リスクの程度など大きく異なるものです。
そのため、多種多様な製品に応じて合理的な規制が必要であるという特性があります。
PMDAでは、これらの医療機器のうちハイリスク医療機器
(例:人工心臓、ペースメーカー、冠動脈ステント、人工血管、人工関節、人工腎臓等)を中心に、
承認審査を行っています。
あとで、説明しますが、クラスⅢ、Ⅳに該当する医療機器です。
医療機器の承認審査では、
このような医療機器の特性を踏まえた上で、
より優れた医療機器をより早く医療現場に提供するため、
審査期間目標を設定し、業務の迅速化に取り組んでいます。
と言っても、米国のFDAに比べると倍以上の期間が掛かります。
また、クラスⅡの医療機器は、
概ね、第三者認証機関に認証を移行しています。
さらに、人に与えるリスクの低い、クラスⅠの医療機器は
登録するだけで販売することができるようになっています。
ただし、勘違いしないで下さい。
登録というのは、
必要な書類は全て揃った上での登録です。
再生医療等製品承認審査業務
平成25年11月27日に公布された医薬品医療機器法において、
再生医療等製品が新たに定義されました。
再生医療等製品は、人や動物の生きた細胞・組織を用いた製品や遺伝子治療用の製品であることから、
従来の医薬品・医療機器と異なる性質を持っています。
例えば、生きた細胞を用いて製造された治療用の製品は、
品質が一定にならないことが多くあります。
しかし、その有効性が推定され、安全性が確認されれば、
条件及び期限付きで特別に早期に承認できる仕組みとして、
「条件及び期限付き承認制度」が導入されました。
これによって、多くの難病の方が救われる可能性が出てきたのです。

ISO13485:2016+QMS省令対応の品質マニュアルのお求めは、上の画像をクリックして下さい。